















































软件

产品


摘要
冷冻电镜(cryo-EM)被广泛用于解析与G蛋白或抑制蛋白复合的活化G蛋白偶联受体(GPCRs)的结构。
然而,将其应用于没有信号蛋白的GPCR仍然具有挑战性,因为大多数受体在其可溶性结构域中缺乏结构特征以促进图像对准。在GPCR晶体学中,于跨膜螺旋5和6之间插入一个融合蛋白是一个非常成功的结晶策略。尽管类似的策略有可能广泛地促进不含信号蛋白的GPCRs的冷冻电镜结构解析,但使这种方法成功的关键决定因素还不清楚。
在这里,我们通过探索不同的融合蛋白设计来解决这一缺陷,这导致了拮抗剂结合的A2A腺苷受体在3.4Å的分辨率和在3.7Å的分辨率下未配位的Smoothened结构。这里探讨的融合策略可能适用于其他GPCR和小型整体膜蛋白的冷冻电镜解析。

| Introduction 简介 |
PCR是最大的整体膜蛋白家族。所有的GPCR都有一个共同的七跨膜螺旋(7TM)结构,并被分为六个家族,从A到F1。激动剂结合后,GPCR通过与异源三聚体G蛋白、G蛋白偶联受体激酶(GRKs)和抑制蛋白2相互作用,激活各种下游信号通路。
作为一个家族,GPCR代表了许多人类疾病的一类重要的药物靶点3。在过去的十年中,方法学的进步为GPCR功能的结构和机制提供了深刻的见解。继续对所有功能状态下的GPCR进行结构研究,包括未配体的apo、被拮抗剂灭活的apo、被激动剂激活的apo以及与下游信号蛋白复合物中的GPCRs的持续结构分析,对于理解GPCR信号转导以及促进治疗发展仍然至关重要。
X射线晶体学最初提供了一种强大的方法来确定许多GPCR结构。虽然像GPCRs这样的完整膜蛋白很难结晶,但一个非常成功的策略是设计一种融合蛋白来取代位于跨膜螺旋5 (TM5)和6 (TM6)之间的第三个细胞内环(ICL3),以便插入的融合蛋白可以介导晶体接触4,5。这种方法与脂质立方相晶体学相结合,已经在RCSB蛋白质数据库中获得了400多个沉积结构。
虽然X射线晶体学在获得与高亲和力拮抗剂或激动剂结合的GPCRs的结构方面非常成功,但获得未配位的受体或GPCR-信号蛋白复合物的结构仍然具有挑战性,这可能是由于未配位或活性状态的受体的结构动力学抑制了晶体形成。
冷冻电镜(cryo-EM)已经成为结构测定的一种革命性的方法。
冷冻电镜的结构测定并不依赖于蛋白质的结晶,而是依靠计算排列和平均单个分子的图像来产生三维(3D)重建6,7。然而,冷冻电镜的一个核心挑战是,三维重建的分辨率取决于单个分子图像的相互对齐的准确性。大多数GPCRs的平均分子量小于40kDa,其关键的结构特征嵌入在膜内;在纯化的系统中,这些区域往往存在于洗涤剂胶束或脂质纳米盘内。对于这种突出于7TM域没有明显可区分结构特征的小膜蛋白,颗粒对准是非常具有挑战性的。
尽管如此,冷冻电镜已经彻底改变了GPCR信号蛋白复合物的结构测定。这一成功的一个关键原因是,刚性连接的信号蛋白驱动粒子在膜内7TM区域的排列(补充图1a)。更广泛地说,对于膜外没有明显结构特征的小膜蛋白靶标,如果刚性结合,添加抗体片段(Fab)等基准标记可以促进精确的图像对齐8。
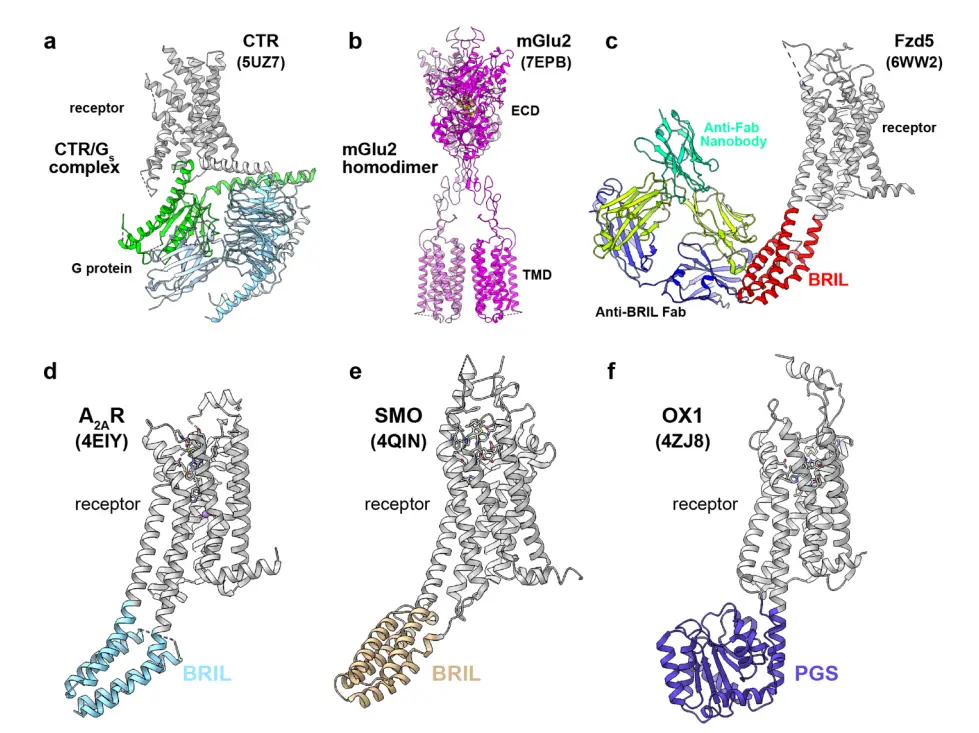
补充图. 1 | 通过单颗粒冷冻电镜确定GPCRs结构的策略 「A. 降钙素受体(CTR)-Gs复合物是通过单颗粒冷冻电镜确定的GPCR/G蛋白复合物的一个例子 B. 谷氨酸受体的结构是通过单颗粒冷冻电镜确定的GPCR二聚体的一个例子 C. hFzd5-Bril与anti-BRIL Fab和anti-Fab nanobody13复合物的冷冻电镜结构。 D-F. 融合蛋白BRIL和PGS插入A2AR-BRIL24(d)、SMO19(e)和OX129(f)中的例子。这些构建体有利于通过X射线晶体学进行结构测定。在这些例子中,BRIL以两个延伸螺旋插入A2AR(d),以一个延伸螺旋插入SMO(e)。PGS被插入一个扩展螺旋(f)」
在理想的情况下,冷冻电镜方法还可以在没有信号蛋白的情况下对GPCRs进行常规结构测定。这将有可能实现对拮抗剂结合的受体或特别是未配体的受体的分析,而且还没有结晶的固有挑战。虽然现在有一些未配体的B族和C族GPCRs的冷冻电镜结构,这些受体在体外形成稳定的二聚体和/或有相对稳定的细胞外结构域(补充图1b)9-11。
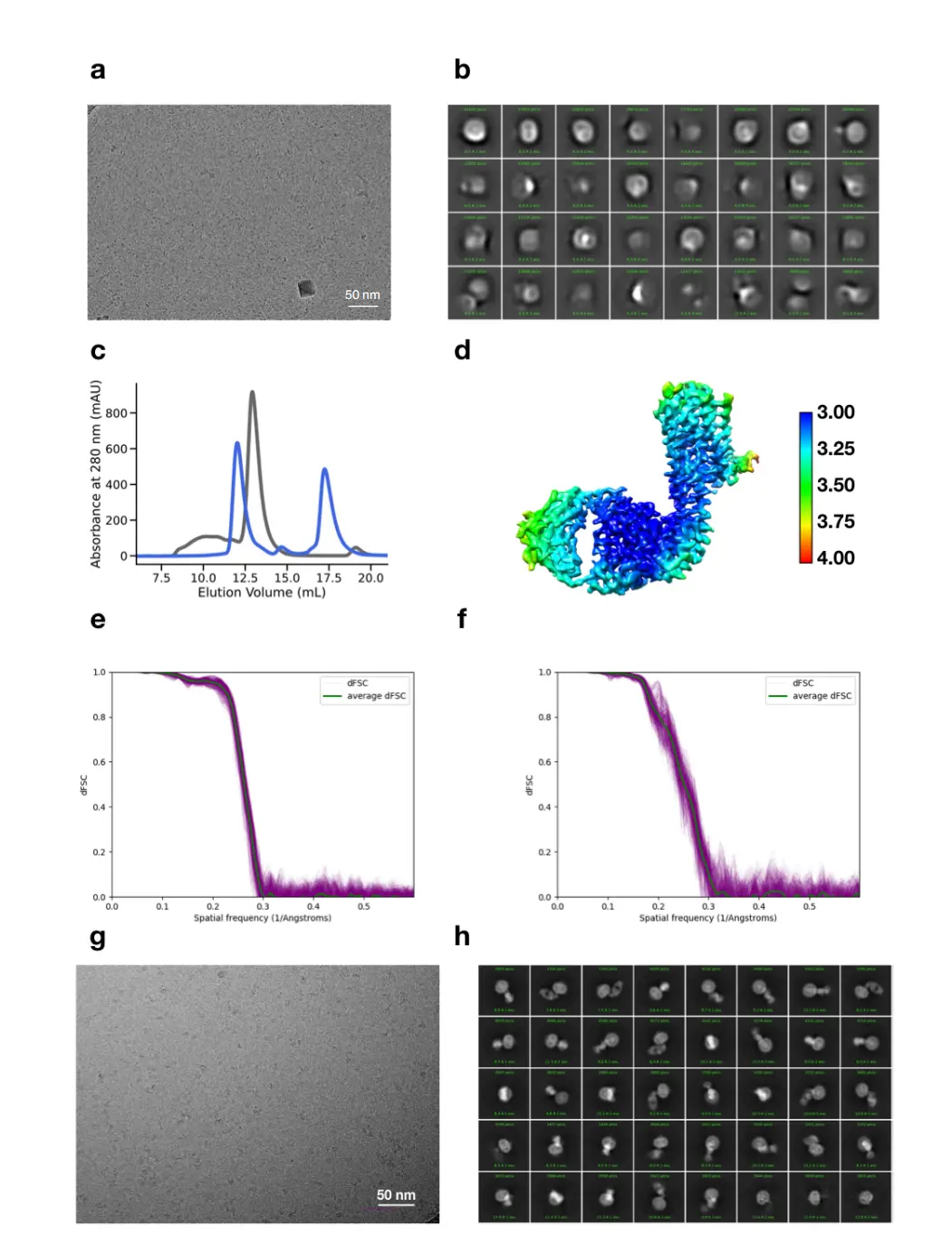
补充图. 2 | A2AR-BRIL的单颗粒分析 「A. A2AR-BRIL样品的代表性原始图像 B. A2AR-BRIL样品的二维平均值 C. 纯化的A2AR-BRIL(灰色)和A2AR-BRIL/Fab复合物(蓝色)的SEC曲线 D. 按局部分辨率着色的密度图 E&F. 完整复合物和TM区域的定向傅里叶壳相关(dFSC)曲线 G. A2ARBRIL/Fab复合物的代表性原始图像。」
最近,纳米抗体已被证明可以通过识别嫁接的细胞内环路来实现非活性状态下的GPCRs的高分辨率重建12。原则上,将融合蛋白刚性地附着在没有大的细胞外结构域的较小GPCR上,可能足以驱动粒子在膜区的排列,产生7TM结构域的高分辨率重建。
事实上,最近在F族人类Frizzled5(hFzd5)受体上的成功表明,ICL3融合apocytochrome b562融合蛋白BRIL,结合抗BRIL Fab片段和抗Fab纳米抗体,可以产生受体7TM结构域的可解释性重建(补充图1c)13。然而,一个基本的限制是,这种成功的重建所需的设计原则仍不清楚。
我们在这项研究中的目标是了解哪些因素能使使用融合蛋白策略的单个GPCR的三维重建取得成功。我们旨在解决两个重要的设计考虑:
(1)成功的粒子对准是否需要有一个特定的融合蛋白尺寸?
(2)融合蛋白通过螺旋延伸的刚性附着对粒子的排列是必需的吗?
我们使用两个模型系统来进行这种分析,即腺苷A2A受体(A2AR)和Smoothened受体(SMO)14-22,这两个受体以前都是通过X射线晶体学和冷冻电镜来分析的。通过探索各种融合策略,我们确定了洗涤剂胶束中拮抗剂结合的无活性A2AR的3.4Å分辨率结构和脂质环境中未配位的无活性SMO的3.7Å结构。这两个例子为融合蛋白的使用提供了设计指南,使得冷冻电镜可能适用于其他GPCRs和其他小的整合膜蛋白。
| Results 成果 |
—「与Fab结合的刚性连接的融合蛋白使A2AR结构得以实现」
我们首先探讨了GPCR融合蛋白的策略是否可以实现7TM区域的高分辨率结构测定。受最近hFzd5的3.7Å分辨率冷冻电镜结构的鼓舞(补充图1c)13,23,我们试图确定融合蛋白策略的"最低要求"。因此,我们使用了以前产生了1.8Å晶体结构的A2AR构建体(补充图1d)24。
这个构建体的几个特点是值得注意的。首先,该结构是用高亲和力拮抗剂ZM241385确定的。其次,该构建体含有热稳定突变,可能进一步限制了构象的异质性。最后,该构建体的晶体结构显示,BRIL结构域通过两个连续的螺旋将受体的TM5和TM6分别连接到BRIL的N端和C端,与受体7TM结构域紧密相连。
这些特点使其成为测试融合蛋白驱动图像对准要求的理想例子。因此,我们在与ZM241385结合的L-MNG/CHS中纯化了A2AR-BRIL融合构建体,进行冷冻电镜研究(补充图2)24。
我们最初试图确定ZM241385与A2AR-BRIL结合的结构,依靠紧密连接的BRIL结构域(MW ~10 kD)作为唯一的参照物。尽管进行了大量的数据收集和图像处理,我们既无法确定具有足够清晰结构特征的二维类平均数(补充图2a,b),也无法生成合理的三维重建,即使我们使用低通滤波的晶体结构作为初始参考模型。
这些结果表明,单独的BRIL结构域,即使牢固地附着在蛋白质上,也不能提供足够的特征来实现GPCR的颗粒排列。按照hFzd5的成功策略,我们接下来添加了一个抗BRIL的Fab片段来扩大靶标。
显然,更大的靶标成功地推动了粒子对准,并产生了一个全局分辨率约为3.4Å的重建。进一步集中细化,将跨膜域的分辨率提高到3.2Å,足以使模型建立(图1,补充图2和3)。

图. 1 | A2AR-BRIL的单颗粒冷冻电镜结构 「A-C. 与抗BRIL Fab结合的A2AR-BRIL的冷冻电镜密度图的三个不同视图 D&E. 结合口袋中化合物ZM241385的特写视图 F. 磷脂酰丝氨酸的对接所强调的脂质密度位置」
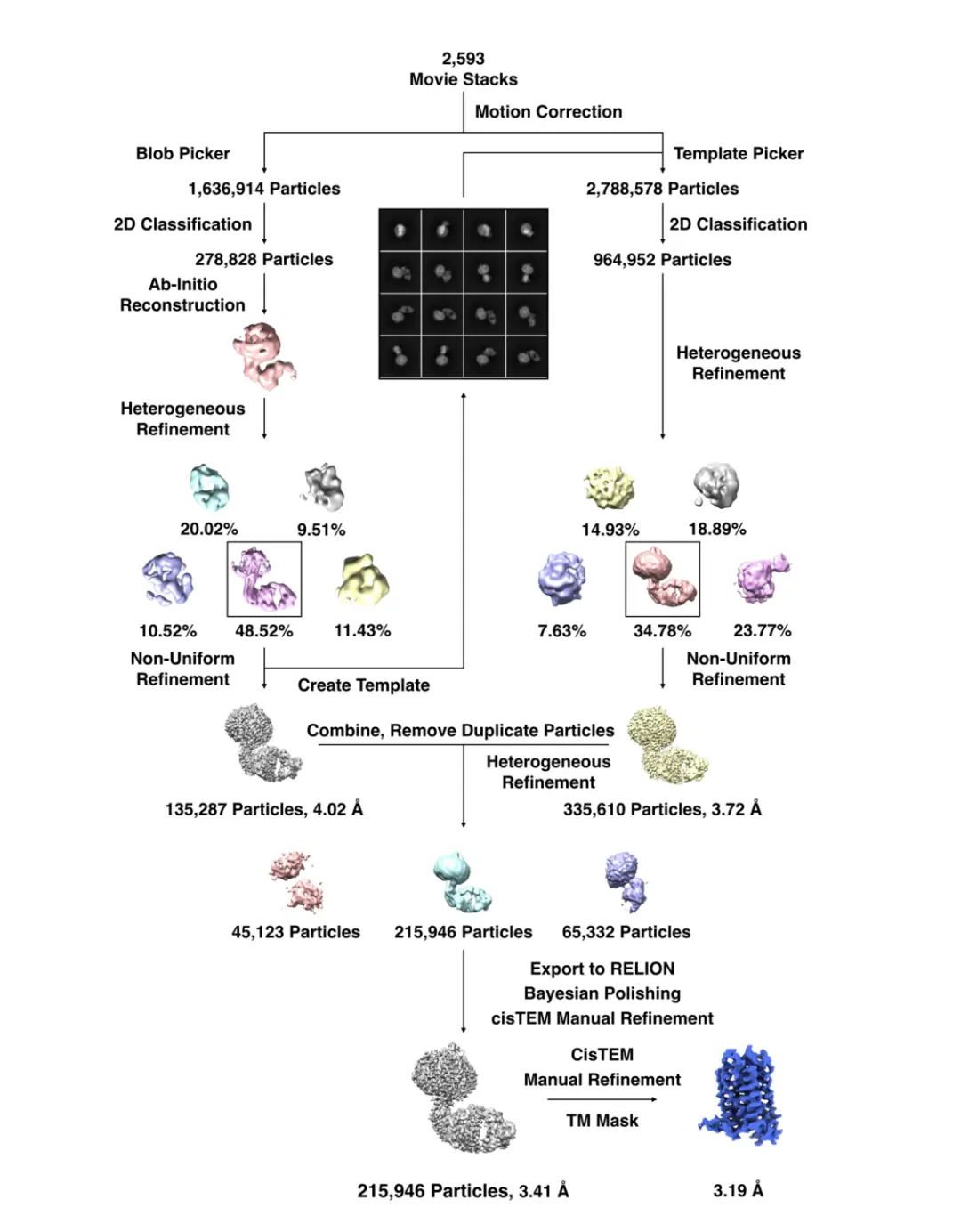
补充图. 3 | A2AR-BRIL/Fab的冷冻电镜数据分析流程图 「流程图说明了确定A2ARBRIL/Fab复合物冷冻电镜结构的数据处理过程」
A2AR-BRIL/Fab的冷冻电镜重建与A2AR-BRIL的X射线晶体结构几乎完全一致,整体均方根偏差(RMSD)为0.9Å。配体ZM241385在配体结合袋中被很好地解析(图1d,e),表明该策略可以使药物结合可视化。
此外,我们在A2AR附近的膜内叶上发现了一个具有两个脂肪族尾巴的脂质密度,以及TM5和TM6之间的一个极性头团,这在蛋白质数据库中以前的58个A2AR的X射线和冷冻电镜结构中都没有观察到。我们推测它是磷脂酰丝氨酸,因为它与密度最匹配(图1f),并且与鉴定磷脂酰丝氨酸为A2AR25共纯化脂质的天然质谱研究一致。
基于这些结果,我们得出结论,Fab与刚性附着的BRIL紧密结合是必要的,以作为单颗粒图像对准的基准标记。
—「单个螺旋BRIL连接不足以进行高分辨率重建」
我们接下来旨在了解是什么因素驱动了融合蛋白与GPCR的紧密连接。对于A2AR和hFzd5,BRIL是以两个延伸的螺旋连接到7TM结构域的。
然而,这种安排要求TM5和TM6的方向与BRIL的N端和C端螺旋精确匹配;事实上,hFzd5的结构需要在这个交界处进行重大的工程设计才能成功13。更常见的是,BRIL结构域仅通过一个延伸的螺旋插入到GPCR或其他靶蛋白中19,因为刚性连接不是结晶的必要条件。
因此,我们问道,当另一个不一定嫁接有刚性或短连接体时,连接到目标蛋白上的融合蛋白只在一个连接体部位有一个延伸的螺旋,是否能促进高分辨率的结构测定。
对于这种方法,我们使用了小鼠SMO(mSMO),它是另一种GPCR,先前已经确定了晶体和冷冻电镜结构。野生型mSMO包含一个7TM结构域和一个细胞外富含半胱氨酸的结构域(CRD)17,18。以前对SMO与Gi结合的冷冻电镜研究无法解决CRD区域,这表明它是灵活的,不太可能驱动图像排列。然而,像A2AR一样,在SMO ICL3中插入一个BRIL结构域促进了SMO的结晶14,19(补充图1e)。
值得注意的是,与A2AR晶体结构不同,该构建体在SMO的TM5和BRIL的N端之间只有一个延伸螺旋。因此,我们使用这种融合策略来测试BRIL与单一扩展螺旋的连接,无论是否有抗BRIL Fab,是否足以驱动图像对准。
早期的研究将脂质纳米盘作为一个受控的膜环境来检测胆固醇对SMO活性的要求26。因此,我们将纯化的mSMO-BRIL重组到由saposin(Salipro)27形成的脂质纳米颗粒中,并加入胆固醇。作为一种膜支架蛋白,saposin允许形成与重组膜蛋白大小相适应的脂质纳米颗粒,提供了脂质环境的好处,但没有给纳米颗粒增加过多的非结构化密度。
一致的是,与没有抗BRIL Fab的A2AR-BRIL的情况一样,我们无法单独确定mSMO-BRIL的高分辨率结构(补充图5a,d)。然而,与A2AR-BRIL/Fab的情况不同,加入抗BRIL Fab以扩大基准标记并没有产生任何改善(补充图5e)。这不太可能是由mSMO重组到saposin纳米颗粒造成的。
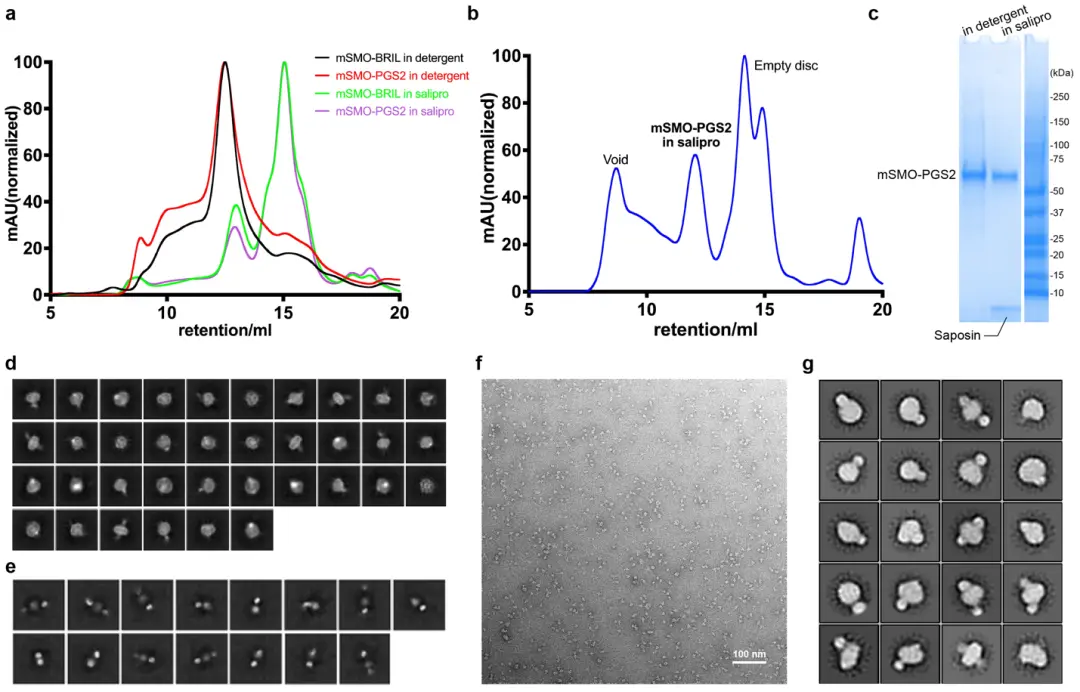
补充图. 5 | 评估SMO-PGS/BRIL的制备 「A. 纯化的mSMO-BRIL和mSMO-PGS2在洗涤剂或salipro中通过凝胶过滤的筛选结果 B. 在制备低温载网之前,mSMO-PGS2在salipro中的凝胶过滤结果 C. 经库马西蓝染色的SDS-PAGE凝胶图像 D&E. 没有(d)或有(e)抗BRIL的mSMO-BRIL通过低温EM的二维平均值 F&G. 电镜负染下,mSMO-PGS2的一张代表性原始显微照片(f)和二维平均数(g)。」
我们推测,要么单独的BRIL不够大,不能驱动图像对准(如上面A2AR-BRIL的情况所示),要么单独的BRIL与单一的延伸螺旋的连接可能不够紧密,而灵活性会因加入抗BRIL Fab而放大。这些结果表明,一个具有两个延伸螺旋的BRIL与一个抗BRIL Fab结合的构建体对于在冷冻电镜重建中获得足够的分辨率可能很重要。
—「通过PGS融合实现脂质纳米盘中apo SMO的结构」
我们转向另一种融合蛋白的方法,以实现无活性apo mSMO的结构测定。
考虑到精确插入具有两个延伸螺旋的融合蛋白的挑战,我们认为通过一个单一的刚性螺旋连接一个更大的融合蛋白可能更容易设计。我们选择了来自Pyrococcus abyssi(PGS)的热稳定糖原合成酶结构域(MW ~20 kD),这是一种融合蛋白,已被用于确定GPCR晶体结构,包括CB1大麻素受体28,以及OX1和OX2奥曲肽受体29(补充图1f)。
我们预计,假设有一个刚性的连接,单独的PGS,比BRIL体积大,但比BRIL/anti-BRIL fab小,可能足以作为一个具有单一延伸螺旋的基准标记。
我们设计了两个不同的构建体,mSMO-PGS1,其中PGS结构域在ICL3的441位之后和TM6的445位之前插入到SMO,以及mSMO-PGS2,其中PGS在TM6中向上插入了一个半螺旋圈(五个氨基酸)(补充图4)。
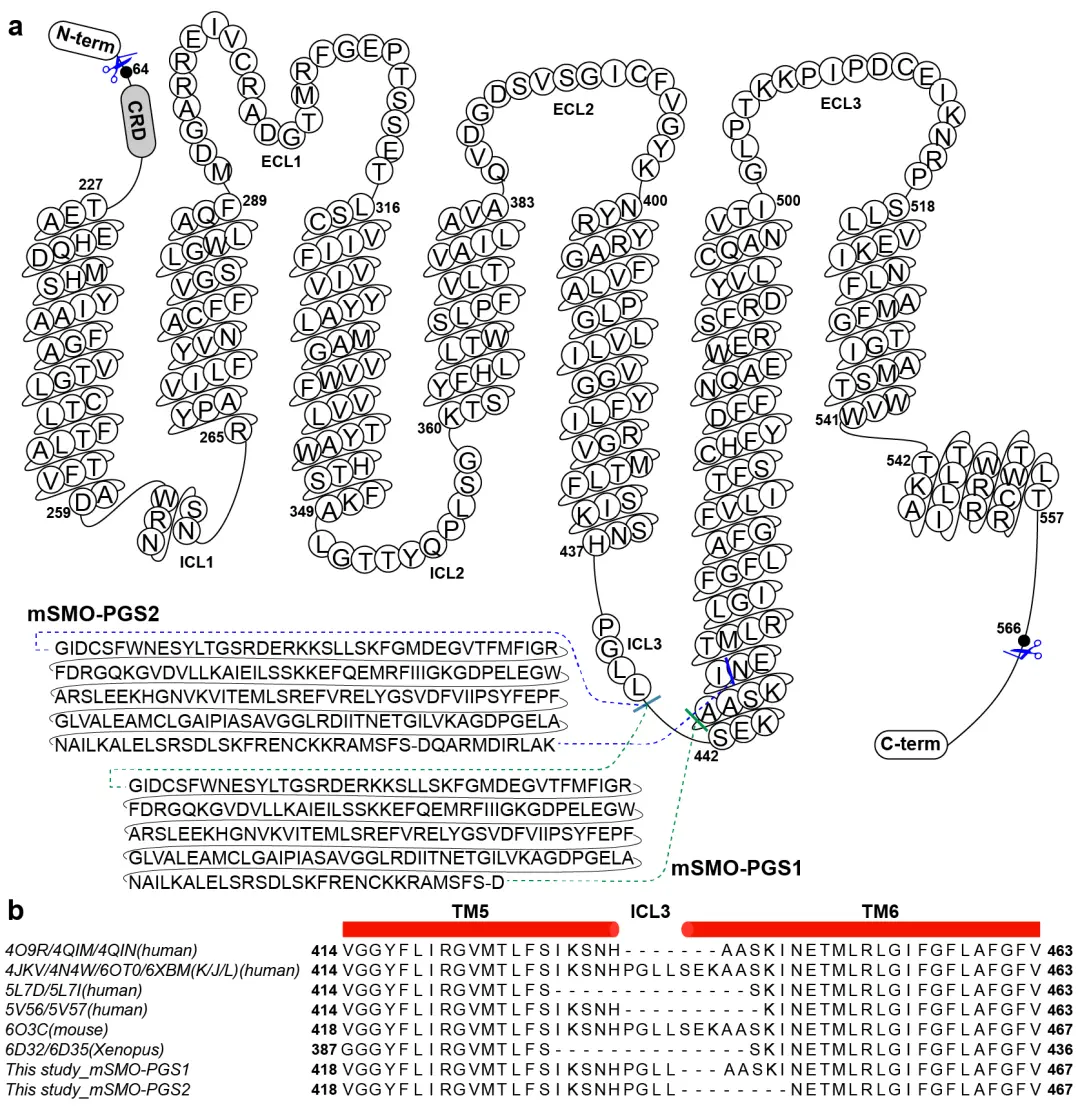
补充图. 4 | SMO-PGS的结构设计 「A. 为SMO-PGS设计的两个构建体的详细图示 B. 本研究和其他出版物中关于ICL3修饰的序列排列。本研究中的mSMO-BRIL构建体与结晶中使用的BRIL插入ICL3相同(PDB ID: 4QIN)」
AlphaFold30预测PGS的C端螺旋在两个构建体中都作为一个连续的螺旋延伸到mSMO的TM6,但在mSMO-PGS1中PGS取向于7TM束的一侧,在mSMO-PGS2中直接位于7TM束的下方(图2a中蓝色和黄色条带)。
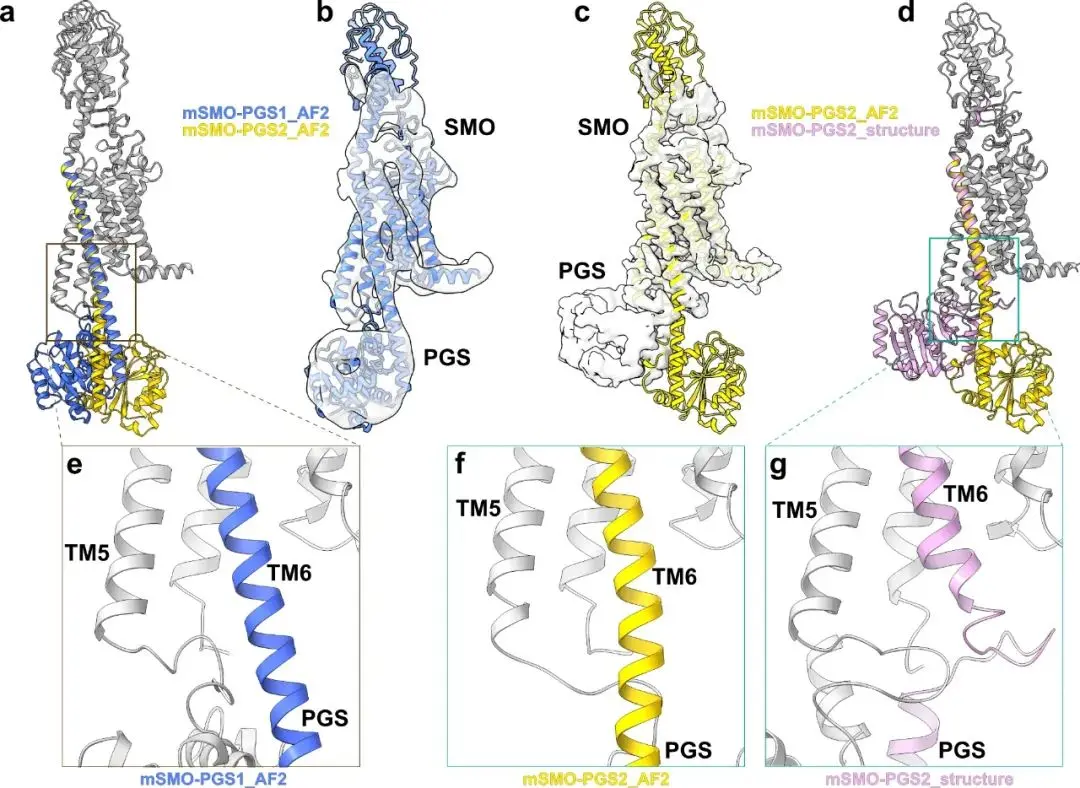
图. 2 | AlphaFold2(AF2)预测的mSMO-PGS1(蓝色)和mSMO-PGS2(黄色)的原子模型。请注意,在这两个构建体中,插入的PGS被预测为在两个相反的方向上定向。 「B. 根据mSMO-PGS1确定的冷冻电镜密度,与AF2预测的原子模型对接(蓝色条带)。 C. 由mSMO-PGS2确定的低温电镜密度,与AF2预测的原子模型对接(黄色条带) D. 确定的结构(粉红色条带)和预测的mSMO-PGS2的原子模型的重叠。 E-G. 在mSMO-PGS1的预测模型(e)、mSMO-PGS2的预测模型(f)和mSMO-PGS2的确定结构(g)中,SMO TM6和PGS之间连接区域的放大图。」
在这两种情况下,PGS的N端通过一个环与TM5的C端相连。同上,我们将纯化的mSMO-PGS重组到添加了胆固醇的脂质saposin nanoparticle27。我们得到了重组在纳米粒子中的表现良好的mSMO-PGS蛋白,并使用尺寸排除色谱法的峰值部分制备了冷冻电镜载网(补充图5b-c,和f-g)。
我们成功地从两个mSMO-PGS构建体中确定了冷冻电镜结构。尽管这些构建体的差异很小,但这些结构的分辨率差异很大(图2,补充图6-8)。

补充图. 6 | 构建体mSMO-PGS1的数据处理的工作流程 「流程图说明了确定mSMOPGS1冷冻电镜结构的数据处理过程。」
我们只从mSMO-PGS1中获得了~6 Å的分辨率(图2b和补充图6),这表明融合蛋白的整体结构与AlphaFold预测一致。虽然不足以建立新的模型,但该分辨率足以使TM束的位置得到确定。TM6和PGS的方向表明,mSMO和PGS之间的连接保持了一个延伸的螺旋,与AlphaFold的预测相匹配。这种类型的连接与PGS连接CB1大麻素受体28、OX1和OX2奥曲肽受体的方式相似29,这些受体的晶体结构显示了这种连接。
我们的结论是,与mSMO-BRIL构建体的情况相似,单螺旋延伸到PGS的融合不足以进行高分辨率的重建。
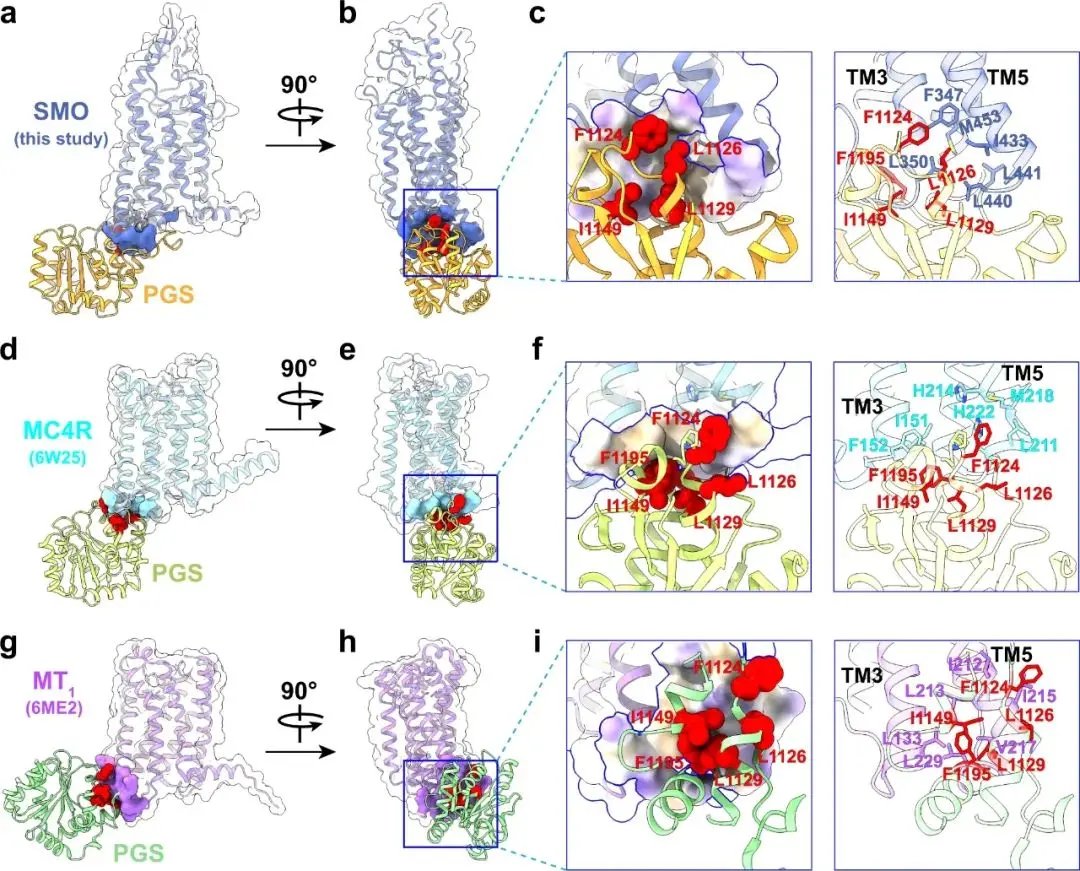
图. 3 | SMO/MC4R/MT1和PGS融合蛋白之间的疏水界面 「A&B. mSMO-PGS2结构的两个不同视图显示了受体和PGS的相对方向 C. mSMO和PGS之间促进PGS刚性附着的疏水相互作用的放大图,包括PGS(左边的球体)和SMO中的关键残基 D&E. MC4R结构的两个不同视图(PDB ID: 6W25 ref. 31) F. 框内区域的放大图显示了在mSMO-PGS2中观察到的相同PGS残基与MC4R中非极性残基的相互作用。i 框内区域的放大图显示了在mSMO-PGS2中观察到的与MT1中非极性残基相同的PGS残基的相互作用。 G&H. MT1结构的两个不同视图(PDB ID: 6ME2 ref. 32)。MT1与PGS的接触区域用海蓝色的表面表示。 I. 框内区域的放大图显示了在mSMO-PGS2中观察到的与MT1中非极性残基相同的PGS残基的相互作用」
令人惊讶的是,mSMO-PGS2的重建与AlphaFold的预测不同,但达到了3.7 Å的明显更好的分辨率(图2c,补充图7和8)。与AlphaFold预测不同的是,PGS位于TM束下面的方向,在mSMO的TM6和PGS的C端之间没有一个连续的螺旋,而是一个扭曲的环,使PGS结构域直接与mSMO 7TM束相互作用。在mSMO和PGS的界面上,PGS的一个疏水环(F1124、L1126、L1129、I1149和F1195)与TM3和TM5底部的疏水残基(F347、L350、I433和L440)接触(图3a-c)。
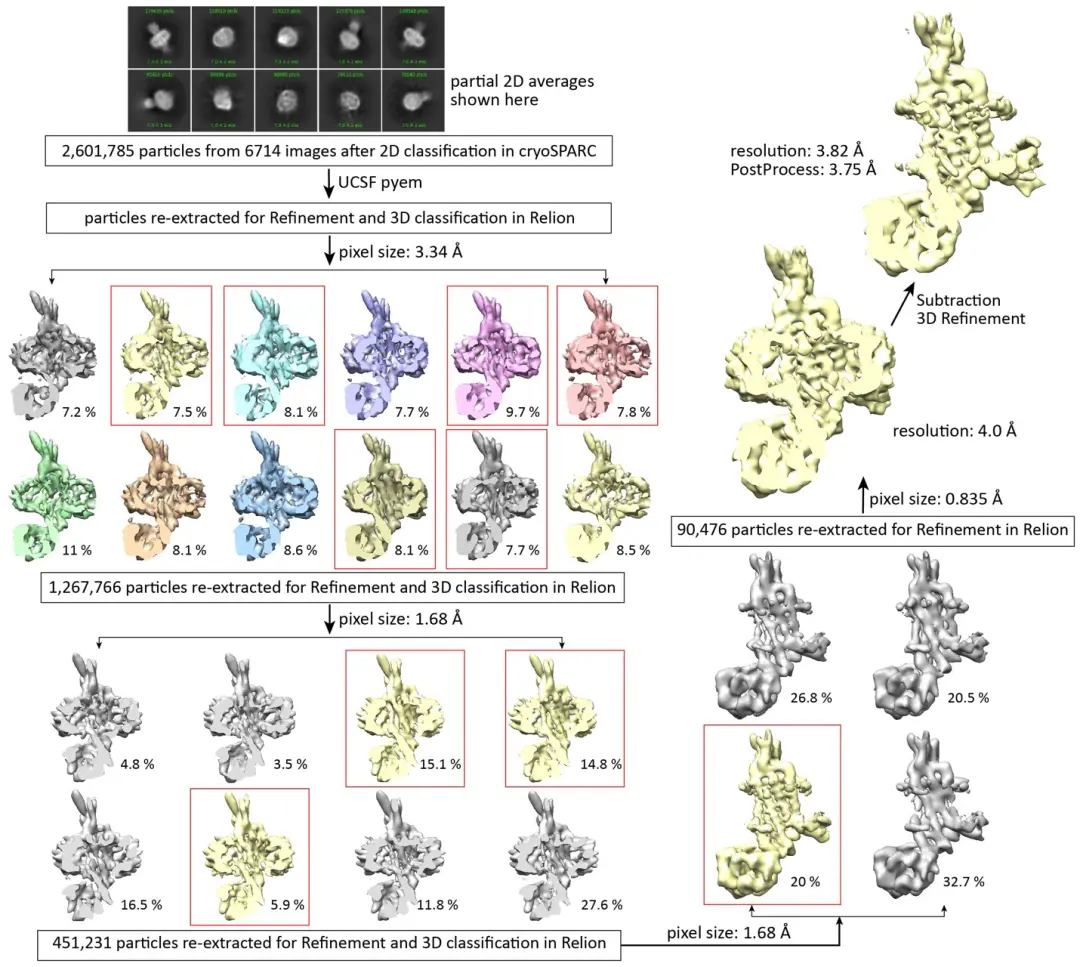
补充图. 7 | 构建体mSMO-PGS2的数据处理的工作流程 「流程图说明了确定mSMOPGS2冷冻电镜结构的数据处理过程。」
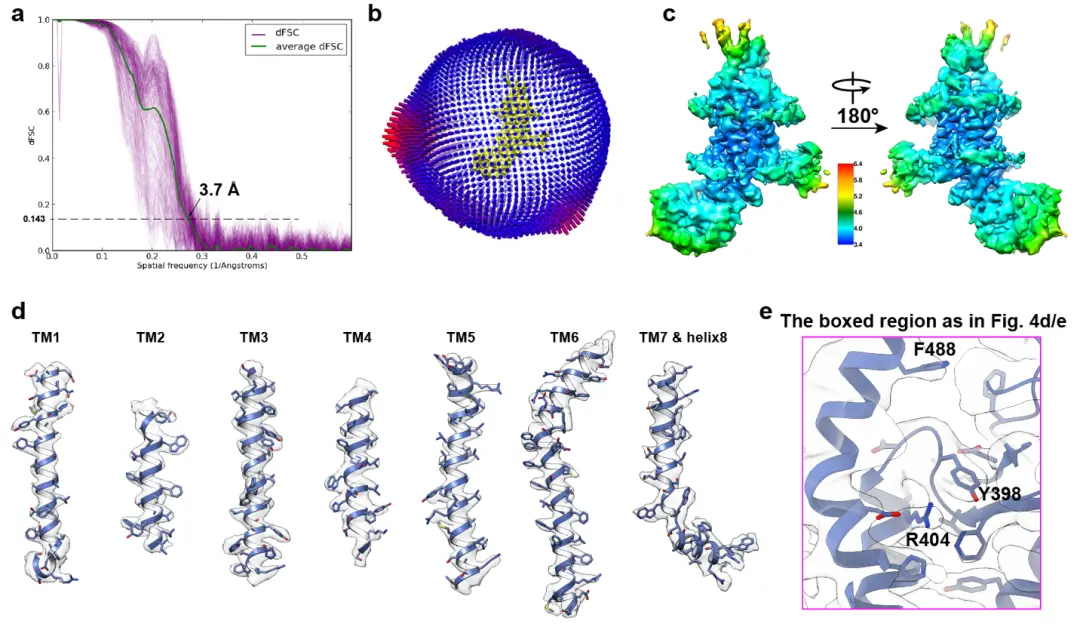
补充图. 8 | 高分辨率下mSMO-PGS2的冷冻电镜结构 「A. 方向性傅里叶壳相关(dFSC)曲线 B. 重建中使用的最终粒子的欧拉角分布的直方图表示 C. 使用局部分辨率(Å)信息从最高分辨率(深蓝色)到最低分辨率(红色)着色的电镜图 D. 跨膜螺旋的密度和原子模型 E. 图.4 d/e中的方框区域的放大图,突出mSMO-PGS2在密度图中的建模。三个残基被标记,包括Y398、R404和F488」
这种与mSMO细胞内侧的未预料到的疏水作用可能稳定了PGS相对于mSMO的方向。我们推测,这种相互作用使得PGS能够刚性地附着在mSMO上,并促进了mSMO 7TM结构域的高分辨率重建。如果PGS结构域作为融合蛋白插入到TM5和TM6的两个非刚性环连接,那么类似的相互作用很可能发生在其他GPCR中。
事实上,我们发现了两个例子,其中融合的PGS促进了MC4R(PDB代码:6W2531)和MT1(PDB代码:6ME232)的结晶。这两个晶体结构显示了PGS结构域和GPCRs之间类似的疏水相互作用(图3d-i),从这三个结构计算出的PGS和GPCRs之间的埋藏面积为836 Å2(mSMO),350 Å2(MC4R),和533 Å2(MT1)。
在H1R和GLP-1R中也可以找到类似的疏水残基(补充图9a,b)。值得注意的是,人类SMO中相当的疏水残基也与SMO-Gi复合物(PDB ID:6XBM)中Gi的α5螺旋中的疏水残基相互作用(补充图9c),表明PGS和Gi与SMO的相互作用是相似的,即使SMO在本研究中处于无配体的非活性状态,但与Gi复合物中处于兴奋剂结合的激活状态。考虑到G蛋白中这些高度保守的疏水残基(补充图9d-f)被认为与其他GPCRs的类似疏水区域相互作用,我们预测PGS与其他GPCRs的TM3/TM5区域之间可能存在类似的相互作用。
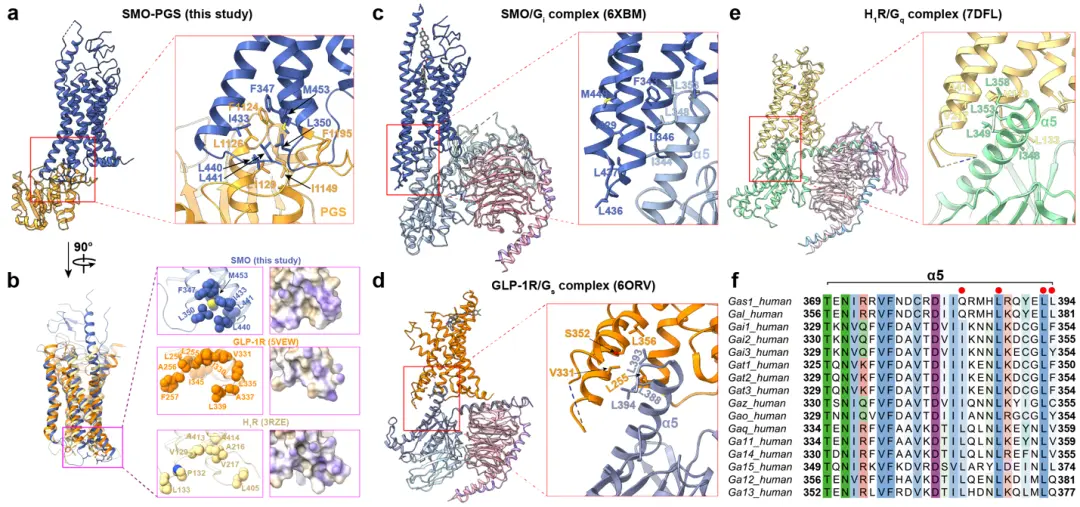
补充图. 9 | SMO-PGS和GPCR/G蛋白复合体中存在的疏水相互作用 「A. SMO和PGS之间的疏水相互作用B. SMO(本研究)与GLP-1R(PDB ID: 5VEW)和H1R(PDB ID: 3RZE)的结构叠加。C. 人类SMO/Gi蛋白复合物的疏水相互作用D. GLP-1R/Gs蛋白复合物的疏水相互作用E. H1R-Gq蛋白复合物的疏水相互作用F. G蛋白家族中α5的序列排列。」
我们在此确定的两个结构,比较表明,PGS作为一个融合蛋白足以促进高分辨率的结构测定,但需要与SMO的稳定连接。与此相关的是,与PGS的单一延伸螺旋连接而没有额外的蛋白(蛋白相互作用)不足以产生所需的结构刚性。
我们早期使用单螺旋的mSMO-BRIL融合的结构上的不确定结果进一步证实了这一结论。相反,在mSMO-PGS2中PGS和mSMO之间的疏水作用提供了足够的稳定,以实现高分辨率的结构(图3c和补充图9a)。
—「脂质环境中apo SMO的结构揭示了一个甾醇部位」
SMO是Hedgehog信号通路的一个关键成分,牵涉到胚胎发育和成人组织稳态33。SMO功能障碍导致出生缺陷和癌症34。SMO的活性由Hedgehog(Hh)的受体Patched调节。
目前一个关于Hedgehog信号的模型表明,Patched通过膜运输甾醇,并在膜内页保持较低的局部甾醇浓度;这使SMO失去活性35。在与Hedgehog结合时对Patched的抑制会增加内页的甾醇浓度并释放SMO的抑制。在迄今发表的17个SMO结构中,有12个受体单独或与纳米抗体复合物的晶体结构和5个受体与Gi蛋白复合物的冷冻电镜结构,尽管Gi蛋白参与SMO信号传导途径仍有争议36。
我们在saposin脂质纳米圆盘中的mSMO的冷冻电镜结构使7TM和连接域(LD)的建模成为可能,它将细胞外CRD与7TM域连接起来(图2c)。
SMO CRD的大部分在我们的图中分辨率很低,除了在连接7TM和CRD的区域有两个小的密度片段,其中一个有短螺旋的外观。apo SMO中CRD的结构灵活性类似于以前与Gi结合的活性SMO的冷冻电镜结构,其中CRD是未解决的。相比之下,以前的SMO单独的晶体结构解决了CRD的问题,这表明这个特定的方向可能是由晶体接触稳定的。
我们将SMO与CRD的三个可用的原子模型对接到我们的密度图中,每个模型代表CRD相对于7TM束的不同方向。短螺旋状的密度与对接的CRD结构域中的任何螺旋都不匹配(补充图10),表明我们的结构中CRD的构象可能与所有其他可用的结构不同,或者说,CRD在没有配体或晶体包装的情况下更灵活。
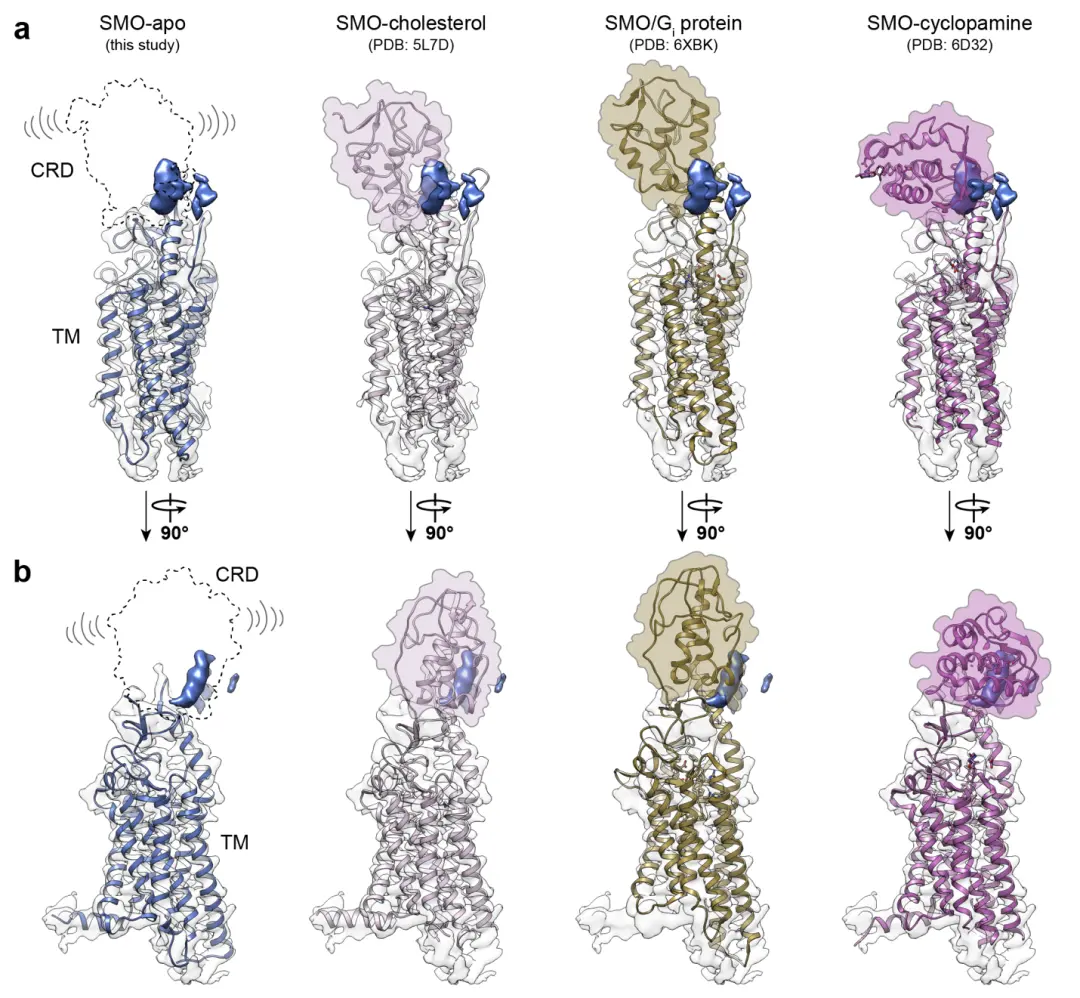
补充图. 10 | 代表性原子模型与mSMO-PGS2密度图的对接 「A&B. 从左到右,对接新建立的原子模型,SMO-胆固醇,SMO/Gi蛋白,和SMO-环胺模型。」
与以前的SMO结构相比,我们的apo SMO结构与非活性SMO相似,7TM区域的整体RMSD为0.75Å,TM5和TM6的构象与非活性受体一致(图4a)。SMO在7TM结构域内有一个大的口袋,这是内源性甾醇的结合点,对通路的激活和合成的小分子兴奋剂、拮抗剂和异生调节剂很重要15,19。与apo状态一致,在我们的结构中,这个口袋是空的,没有任何结合的配体或甾醇的明显密度(图4b)。事实上,apo SMO的配体结合口袋太窄,无法容纳7TM束内的甾醇,这在以前的一个活性SMO结构(PDB ID:6O3C)15中观察到(图4b,c)。

图. 4 | apo状态的mSMO的结构 「A. apo状态下的mSMO(本研究)与拮抗剂结合的非活性状态(PDB ID:5L7D)和激动剂结合的活性状态(PDB ID:6O3C)结构的比较 B. 根据本研究确定的apo状态结构计算的配体结合袋 C. 相比之下,活性状态下SMO的配体结合口袋(PDB ID: 6O3C)与激动剂SAG21k结合,口袋底部有一个胆固醇 D. 本研究确定的SMO apo结构的原子模型(蓝丝带)与以前确定的(PDB ID: 5L7D)重叠,没有配体但CRD中结合有胆固醇。两者之间的明显差异表明,虽然两个结构都是apo状态,但它们可能呈现apo SMO的两种不同的中间状态 E. b、c所示结构的叠加。 F. 位于mSMO内侧小叶的胆固醇的密度 G. 通过表面视图强调的甾醇位点 H. 与g中看到的观点相同,显示从非活性状态到活性状态的构象变化对甾醇结合的影响」
我们将我们的脂质包埋apo SMO的结构与先前确定的使用脂质立方相方法解出的未配位SMO的X射线晶体结构(PDB ID:5L7D)14进行了比较。
这个先前的结构解决了CRD与胆固醇结合的问题,但在7TM内没有明确的胆固醇密度。与这个结构和SMO的活性状态结构(PDB ID:6O3C)相比,我们发现7TM区域有细微但重要的差异。首先,7TM口袋中的几个残基重新排列,包括Y398(人类SMO(hSMO)中的Y394)、R404(hSMO中的R400)和F488(hSMO中的F484)(图4d,e和补充图8e)。这导致了较小的口袋体积(1630 vs 2292 Å3)。第二,我们发现在7TM束外有一个与TM5和TM6相邻的密度,面向质膜的内叶,其形状和大小与甾醇一致(图4f)。
分辨率排除了特定甾醇的分配,并且不清楚该密度是代表内源性甾醇还是在saposin重建过程中添加的胆固醇。然而,鉴于在我们的重组中使用了高浓度的胆固醇,我们暂且在这个部位建立了胆固醇模型。据我们所知,以前在SMO的X射线或冷冻电镜结构中没有观察到类似部位的脂质密度。模型中的胆固醇结合在由TM5的残基L423和V427以及TM6的L454和F461形成的疏水缝隙中。TM5中残基K434的侧链可能与胆固醇的羟基相互作用(图4f)。值得注意的是,F461(hSMO中的F457)重新定向以适应胆固醇在该部位的结合(图4g)。与SMO的活性状态结构比较表明,这个缝隙在受体激活时被破坏(图4a和h),表明这个部位只存在于非活性或预活性状态15。最后,胆固醇的异辛基尾巴面对着脂质双层的内页和活性SMO的7TM口袋之间的隧道开口(图4a,c)。
这些结构观察使我们推测,甾醇在该部位的结合可能对Hedgehog途径的功能很重要。我们提出,胆固醇在通路激活时最初在SMO的这个部位结合。TM5和TM6的进一步构象变化将胆固醇挤出该部位,这样它就可以翻转并通过进入TM5和TM6之间的开放隧道到达TM束内的胆固醇结合部位。
虽然是推测性的,但我们的建议扩展了新出现的模型,即小叶内侧胆固醇水平受Patched-1的调节,而途径的激活导致小叶内侧胆固醇的增加,并被SMO所感知15,16,17,35。
| Discussion 讨论 |
冷冻电镜在GPCR结构生物学中的应用大大加快了GPCR信号蛋白复合物结构的确定。
然而,由于无法对准单个颗粒,确定非复合物受体的结构仍然很困难。从融合蛋白在晶体学中的应用和通过冷冻电镜确定hFzd5的结构中得到启发,我们开始审视现有的策略,并为GPCR冷冻电镜结构建立新的融合策略。
值得一提的是,在GPCRs中加入融合蛋白的目的只是为了提供一个基准标记,以方便图像对准,而不是为了减少蛋白质的动态或提高稳定性,这也影响了冷冻电镜结构的可实现的分辨率。因此,我们把重点放在通过突变或拮抗剂稳定的GPCRs上。
我们首先在两个模型GPCRs,A2AR和SMO上测试BRIL-融合策略。先前解决的BRIL融合构建体的X射线晶体学结构显示,A2AR-BRIL在受体和BRIL之间形成两个连续的螺旋,而SMO-BRIL只形成一个连续的螺旋。对于A2AR,使用抗BRIL的Fab使图像对准产生了受体及其药物结合袋的高分辨率重建。然而,在没有Fab的情况下,粒子对准失败。对于SMO,无论是单独的BRIL融合还是BRIL融合加上抗BRIL Fab,都无法实现高分辨率的重建。
综上所述,我们的结果表明,对于没有大型稳定的细胞外结构域的GPCRs,BRIL-融合方法既需要BRIL和受体之间的刚性连接物,如具有两个连续的螺旋或一个由另一个优化的连接物支持的连续螺旋,如EBI2的最新研究中所证明的37,38,还需要一个额外的Fab来扩大基准标记的尺寸。
在确定了BRIL-融合策略的制约因素后,我们试图测试一种新的融合策略,该策略对螺旋形融合点的内在制约较少,并使用不同的靶标。我们选择将PGS纳入mSMO的ICL3中,假设单一连续螺旋的结构和PGS的大小足以促进图像对准。
令人惊讶的是,我们了解到单一的连续螺旋仍是不够的。相反,没有螺旋特性的延伸允许SMO和PGS之间有稳定的疏水接触,这独立地使粒子对准和最终重建SMO 7TM结构域达到3.7Å的分辨率。这一发现提供了一种通过冷冻电镜确定GPCR结构的替代策略。
我们的A2AR和mSMO的结构提供了关于脂质如何与这两种受体相互作用的结构见解。对于A2AR,我们的冷冻电镜结构显示了与TM5和TM6之间的受体结合的推定的磷脂酰丝氨酸。这种脂质在以前解决的任何A2AR的X射线结构中都没有被观察到。对于SMO,我们在脂质环境中的非活性受体的结构显示了与以前发表的X射线晶体和冷冻电镜结构细微但重要的构象差异。此外,该结构揭示了TM5和TM6之间的甾醇结合位点。这两个案例突出了冷冻电镜在阐明GPCR结构生物学方面的潜在效用。
除了最近的纳米抗体策略,通过识别嫁接的细胞内环12,我们的研究强调了GPCR和融合蛋白之间的刚性耦合对于冷冻电镜结构测定的重要性。我们展示了两种替代方法来实现这种刚性。
一种是将融合蛋白连接到具有两个延伸螺旋的GPCR上,通常是TM5和TM6。在连接GPCR和融合蛋白的一个连接点上的单一延伸螺旋不可能产生足够的刚性来驱动图像对准以进行高分辨率的结构测定,特别是当另一个连接点不是刚性的而是长的和/或灵活的。如果TM5和TM6都能作为延伸的螺旋体直接连接到融合蛋白上,就像A2AR-BRIL的情况一样,插入物可能有足够的刚性来进行结构测定。AlphaFold结构预测可能会进一步促成这种设计。
另一种意想不到的方法是,融合蛋白可以通过其他特异性相互作用与GPCR稳定地相互作用,如PGS和mSMO之间的疏水作用。在这种情况下,仅PGS的大小就足以驱动图像排列。值得注意的是,mSMO中与PGS相互作用的疏水残基在许多其他GPCRs中是保守的,这表明PGS融合蛋白可以更广泛地用于其他GPCRs的结构研究。有趣的是,mSMO-PGS1和mSMO-PGS2代表了PGS融合域相对于GPCRs TM域的两种构型,这两种构型都出现在以前发表的多个GPCRs晶体结构中28,29,31,32。
我们在此提出的研究表明,两者中只有一个可以促进图像的排列。因此,当把这种方法应用于其他GPCRs时,有必要尝试多种连接物的设计以获得所需的连接物。因此,我们对融合蛋白策略的探索使没有共同复合信号蛋白的GPCR的结构测定成为可能,从而为理解GPCR结构、功能和最终的药物设计开辟了新的途径。
| Methods 方法 |
此部分从略
| Reference 相关文献 |
1. Rosenbaum, D. M., Rasmussen, S. G. & Kobilka, B. K. The structure and function of G-protein-coupled receptors. Nature 459, 356–363 (2009).
2. Gurevich, V. V. & Gurevich, E. V. GPCR signaling regulation: the role of GRKs and arrestins. Front. Pharmacol. 10, 125 (2019).
3. Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B. & Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017).
4. Chun, E. et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976 (2012).
5. Rosenbaum, D. M. et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266–1273 (2007).
6. Cheng, Y. Single-particle Cryo-EM at crystallographic resolution. Cell 161, 450–457 (2015).
7. Cheng, Y., Grigorieff, N., Penczek, P. A. & Walz, T. A primer to singleparticle cryo-electron microscopy. Cell 161, 438–449 (2015).
8. Wu, S. et al. Fabs enable single particle cryoEM studies of small proteins. Structure 20, 582–592 (2012).
9. Du, J. et al. Structures of human mGlu2 and mGlu7 homo- and heterodimers. Nature 594, 589–593 (2021).
10. Josephs, T. M. et al. Structure and dynamics of the CGRP receptor in apo and peptide-bound forms. Science https://doi.org/10.1126/ science.abf7258 (2021).
11. Koehl, A. et al. Structural insights into the activation of metabotropic glutamate receptors. Nature 566, 79–84 (2019).
12. Robertson, M. J. et al. Structure determination of inactive-state GPCRs with a universal nanobody. bioRxiv https://doi.org/10.1101/ 2021.11.02.466983 (2022).
13. Tsutsumi, N. et al. Structure of human Frizzled5 by fiducialassisted cryo-EM supports a heterodimeric mechanism of canonical Wnt signaling. elife https://doi.org/10.7554/eLife. 58464 (2020).
14. Byrne, E. F. X. et al. Structural basis of Smoothened regulation by its extracellular domains. Nature 535, 517–522 (2016).
15. Deshpande, I. et al. Smoothened stimulation by membrane sterols drives Hedgehog pathway activity. Nature 571, 284–288 (2019).
16. Huang, P. et al. Structural basis of smoothened activation in hedgehog signaling. Cell 174, 312–324.e316 (2018).
17. Qi, X., Friedberg, L., De Bose-Boyd, R., Long, T. & Li, X. Sterols in an intramolecular channel of Smoothened mediate Hedgehog signaling. Nat. Chem. Biol. 16, 1368–1375 (2020).
18. Qi, X. et al. Cryo-EM structure of oxysterol-bound human Smoothened coupled to a heterotrimeric Gi. Nature 571, 279–283 (2019).
19. Wang, C. et al. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 5, 4355 (2014).
20. Wang, C. et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature 497, 338–343 (2013).
21. Weierstall, U. et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 5, 3309 (2014).
22. Zhang, X. et al. Crystal structure of a multi-domain human smoothened receptor in complex with a super stabilizing ligand. Nat. Commun. 8, 15383 (2017).
23. Mukherjee, S. et al. Synthetic antibodies against BRIL as universal fiducial marks for single-particle cryoEM structure determination of membrane proteins. Nat. Commun. 11, 1598 (2020).
24. Liu, W. et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236 (2012).
25. Yen, H. Y. et al. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 559, 423–427 (2018).
26. Myers, B. R., Neahring, L., Zhang, Y., Roberts, K. J. & Beachy, P. A. Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proc. Natl Acad. Sci. USA 114, E11141–E11150 (2017).
27. Frauenfeld, J. et al. A saposin-lipoprotein nanoparticle system for membrane proteins. Nat. Methods 13, 345–351 (2016).
28. Shao, Z. et al. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 540, 602–606 (2016).
29. Yin, J. et al. Structure and ligand-binding mechanism of the human OX1 and OX2 orexin receptors. Nat. Struct. Mol. Biol. 23, 293–299 (2016).
30. Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
31. Yu, J. et al. Determination of the melanocortin-4 receptor structure identifies Ca(2+) as a cofactor for ligand binding. Science 368, 428–433 (2020).
32. Stauch, B. et al. Structural basis of ligand recognition at the human MT1 melatonin receptor. Nature 569, 284–288 (2019).
33. Khaliullina, H., Bilgin, M., Sampaio, J. L., Shevchenko, A. & Eaton, S. Endocannabinoids are conserved inhibitors of the Hedgehog pathway. Proc. Natl Acad. Sci. USA 112, 3415–3420 (2015).
34. Niyaz, M., Khan, M. S. & Mudassar, S. Hedgehog signaling: an Achilles’ Heel in cancer. Transl. Oncol. 12, 1334–1344 (2019).
35. Zhang, Y. et al. Structural basis for cholesterol transport-like activity of the hedgehog receptor patched. Cell 175, 1352–1364.e1314 (2018).
36. Arveseth, C. D. et al. Smoothened transduces Hedgehog signals via activity-dependent sequestration of PKA catalytic subunits. PLoS Biol. 19, e3001191 (2021).
37. Chen, H., Huang, W. & Li, X. Structures of oxysterol sensor EBI2/ GPR183, a key regulator of the immune response. Structure https:// doi.org/10.1016/j.str.2022.04.006 (2022).
38. Liang, Y. L. et al. Phase-plate cryo-EM structure of a class B GPCRG-protein complex. Nature 546, 118–123 (2017).
39. Gao, Y., Cao, E., Julius, D. & Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351 (2016).
40. Ohi, M., Li, Y., Cheng, Y. & Walz, T. Negative staining and image classification—powerful tools in modern electron microscopy. Biol. Proced. Online 6, 23–34 (2004).
41. Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
42. Zheng, S. Q. et al. MotionCor2: anisotropic correction of beaminduced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
43. Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
44. Dang, S. et al. Cryo-EM structures of the TMEM16A calciumactivated chloride channel. Nature 552, 426–429 (2017).
45. Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in singleparticle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003).
46. Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput Chem. 25, 1605–1612 (2004).
47. Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501 (2010).
48. Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
49. Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 74, 519–530 (2018).
50. Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
51. Berman, H., Henrick, K. & Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 10, 980 (2003).
| 词表 |
1. 激动剂
一种通过增加受体活性以产生生物反应的物质。
2. 拮抗剂(也称中性拮抗剂)
一种能阻断激动剂或反向激动剂的物质,在不存在激动剂或反向激动剂的情况下便没有活性
3. 生物制药
在生物活体中制造的药物,可能含有重组蛋白、糖类、基因疗法或核酸。
4. 电子密度图
电子密度与晶体中每一个晶胞位置的关系图,以二维或三维表示,由解析X射线衍射图案得出。
5. 静电势图(即库仑势图)
即样品中电荷分布的二维或三维表示。在电子显微镜中,静电势图由电子被与样品相互作用时产生的库仑力散射而产生。
6. 反向激动剂
一种通过减少受体的基础活性以产生生物反应的物质。
7. 脂质立方相结晶
基于脂立方相的蛋白结晶技术,采用脂立方相模拟生物膜环境,膜蛋白可以在脂质双分子层中相互接触,在合适的条件下形成晶体。
8. 脂质纳米盘
一个纽扣电池形状的盘状脂质双层,由两个环绕的两亲性螺旋蛋白(膜支架蛋白)稳定并使其可溶于水。
9. 冷冻电镜负染
一种将重金属盐染色剂嵌入并固定在生物标本上,并在室温下进行电子显微镜成像的技术方法。尽管只能在低分辨率(~2纳米)下观察标本的形状,但这种技术对于简单和快速评估样品质量是很有价值的。
水木未来丨视界 iss. 25
武汉格发信息技术有限公司,格发许可优化管理系统可以帮你评估贵公司软件许可的真实需求,再低成本合规性管理软件许可,帮助贵司提高软件投资回报率,为软件采购、使用提供科学决策依据。支持的软件有: CAD,CAE,PDM,PLM,Catia,Ugnx, AutoCAD, Pro/E, Solidworks 等。
 技术文档
技术文档
 推荐好文
推荐好文






 155-2731-8020
155-2731-8020




















