















































软件

产品


Wannier90是一款强大的开源代码,可以生成局域wannier函数,并使用它们来高效精确地计算材料的电子性质。通过wannier90,我们可以获取基于wannier function basis的哈密顿量矩阵,并将其利用于紧束缚模型近似的计算当中。但是程序默认输出的文件内容比较庞大,可以通过设计一些简单的程序帮助我们筛选想要获取的重要信息。这里,本文将使用MATALB脚本(要求版本号大于R2019a)来提取指定多个格点的全部哈密顿量,并将它们各自保存成表格形式,大大方便了数据的处理过程。

先贴出代码(因为目前B站代码块没有对应matlab规则,所以高亮看起来很乱):
clc;clear;
[filename, pathname] = uigetfile('*.dat','Please select wannier90_hr.dat');
pathfile=fullfile(pathname, filename); % Obtain file location
S = importdata(pathfile);
Data = S.data;
Nw = Data(1);Data(1) = [];
Nrpts = Data(1);Data(1) = [];
Ndegen = Data(1:Nrpts)';Data(1:Nrpts)=[];
H = reshape(Data,7,[])';
Direct = [0 0 0; 0 0 1; 1 0 0; 0 1 0; 1 0 1; 1 1 0; 0 0 2]; %Obtain the hopping term on site (l,m,n) as you want
for i = 1:length(Direct(:,1))
l=Direct(i,1);m=Direct(i,2);n=Direct(i,3);
temp= find(H(:,1)==l & H(:,2)==m & H(:,3)==n);
Htemp= H(min(temp):max(temp),:);
Htemp(:,6:7)=Htemp(:,6:7)/Ndegen(max(temp)/Nw^2);
Hmat = reshape(Htemp(:,6),Nw,[]); %replace 6 with 7 to output Im(H)
name = [pathname,num2str([l m n]),'.xlsx']; %Modify '.xlsx' to change the format of output
writematrix(Hmat,name,'WriteMode','replacefile','UseExcel',true);
%If you are not going to output in spreadsheet,
%remember to remove comma-separated pair 'UseExcel',true
end整个代码里需要手动改的只有一项就是Direct矩阵,它是以3*n的形式保存的。若你想输出格点(l,m,n)的哈密顿量,就请把l,m,n以一行的形式写入到Direct里,要输出多个格点就继续增加更多的行。
接下来我们以钙钛矿结构的SrVO3(SVO)为例来具体展示下程序的使用方法。(例子中用到的文件
链接:https://pan.baidu.com/s/16B4q5K07T_2tIvMsVvnC_A
提取码:ks3s )
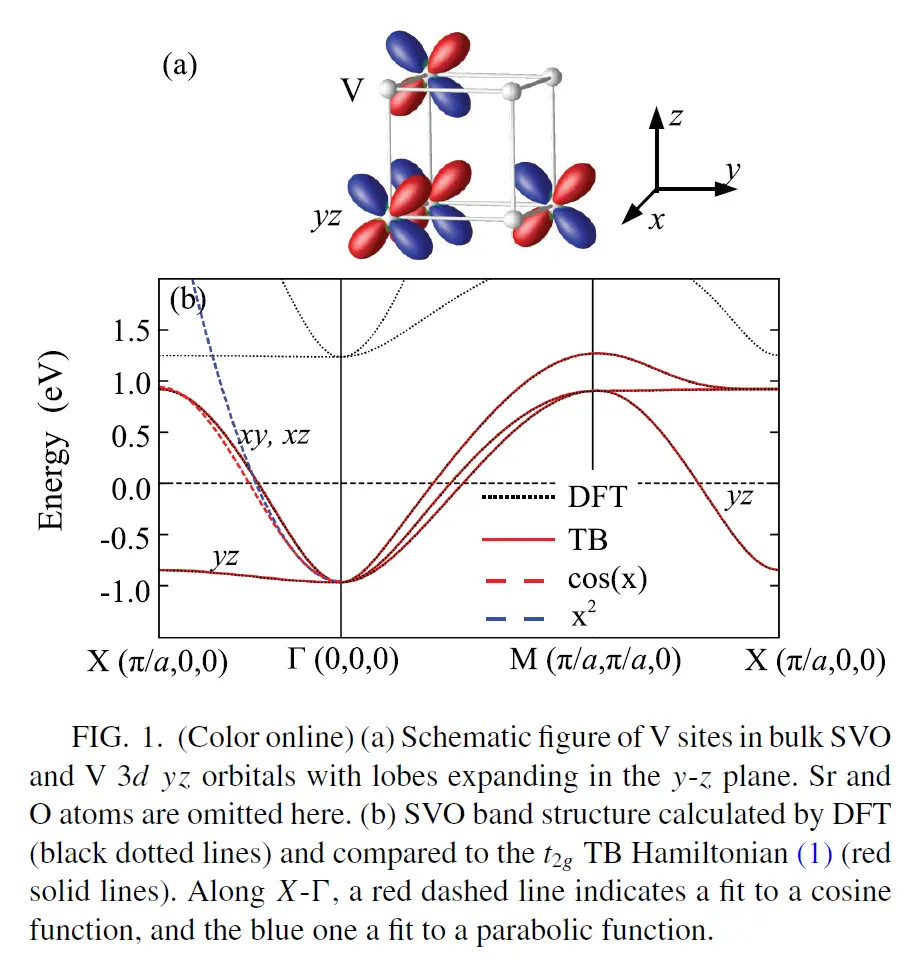
通过DFT求得块体SVO的部分能带如下图所示
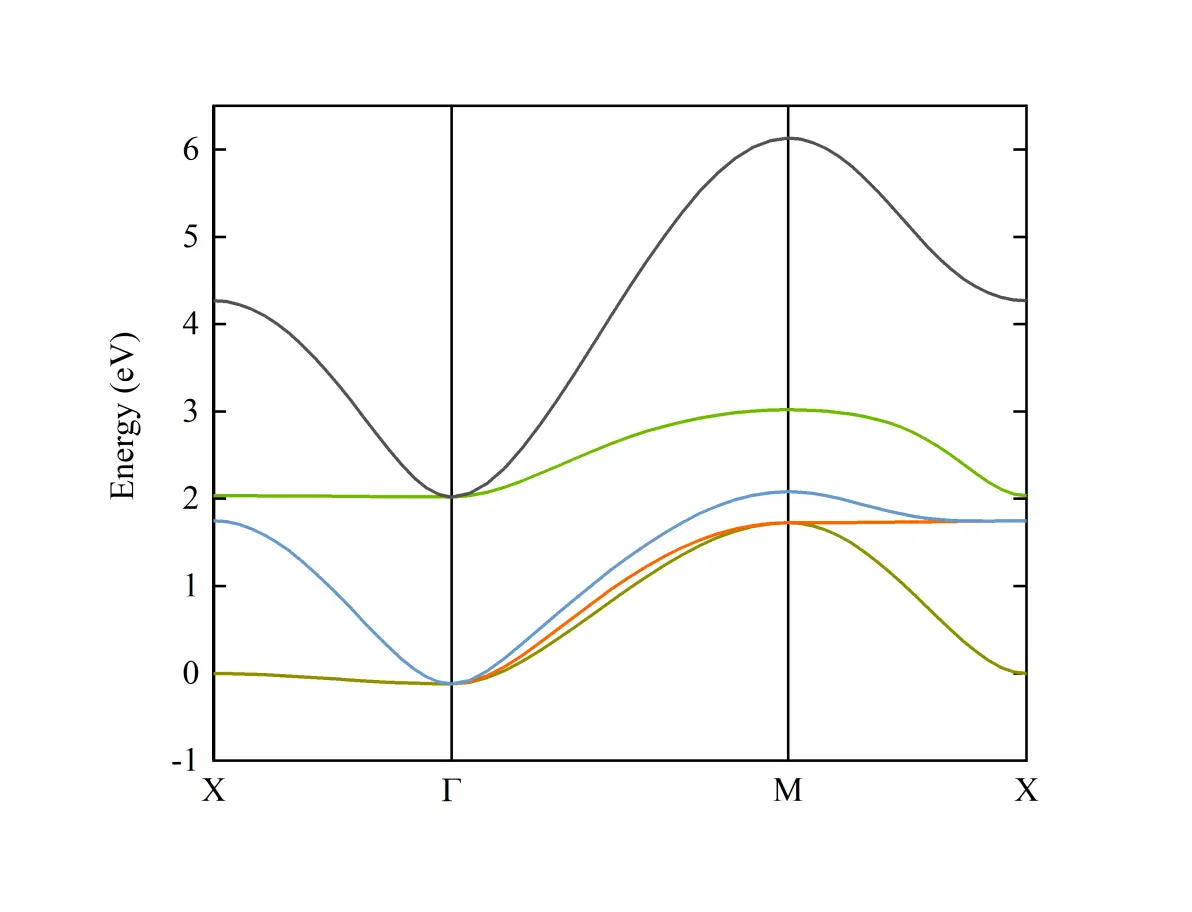
SVO V 3d轨道能带
SVO中V原子的3d轨道能带分布在[0, 6]eV范围内,且与其他轨道没有杂化,非常适合用于做wannier projection。选取V的3d轨道Wannier Function作为Basis,求得wannier90_hr.dat,这一步的具体过程可以参考下站内的一个教程。
建议将wannier90_hr.dat单独保存在一个文件夹内,然后运行程序,此时会弹出一个文件选择界面,选择想要处理的文件并确认。
之后,程序会自动根据你输入的Direct输出对应的哈密顿量并保存为表格形式(表格会根据格点矢量自动命名,并保存在和.dat一个目录下)。如果想保存成文本格式,可以在第19行修改后缀名为'.txt',并移除第20行的最后一个逗号分割对。(PS:如果你很想输出虚部的结果,可以把第18行的6改为7)
简单介绍下下输出的矩阵形式到底是怎样的,以(0,0,0)为例
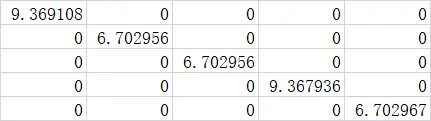
hopping term (0,0,0)
wannier的输出顺序为,l相同时依次输出m=0,±1,±2,...,±l的线性组合。在本文的这个案例中V的3d轨道电子l=2,矩阵从左至右,从上到下每个对应的轨道分别是dz2(m=0),dxz,dyz(m=±1),dx2-y2,dxy(m=±2)。可以看出各轨道电子的格点能是正交化的,且分别对应三重和二重简并的t2g和eg轨道。
接下来这一部分简单谈一下结果分析的部分,有些部分的数学和物理证明会被掠过,如果不太熟悉的朋友可以回头再看一下紧束缚近似的理论。
在这个例子中分析所有的hopping terms,我们可以发现两个关于它们的鲜明特性:
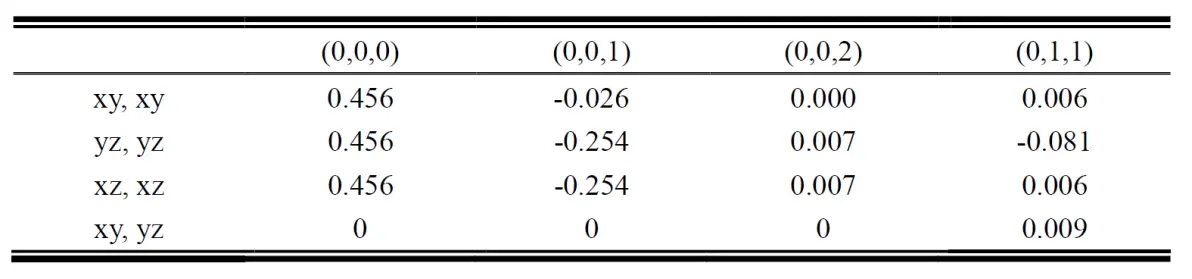
Table I : Hopping integrals in SVO bulk, (0,0,0) have been substracted by fermi energy 6.2472 eV
因此,结合SVO的晶格对称性(cubic),对于每个轨道α沿着特定方向比如z,哈密顿量可以简化为

接下来求哈密顿量中的两个常数ε和t,例如xy轨道沿着z方向,另外根据对称性可得主要的hopping terms 只有四种(建议仔细观察下各个格点的hopping terms,重复的值很多):t0=(0,0,0)=0.456 eV, t1=(1,0,0)=-0.254 eV,t2=(0,0,1)=-0.026 eV, t3=(1,1,0)=-0.081。各自的ε和t分别为
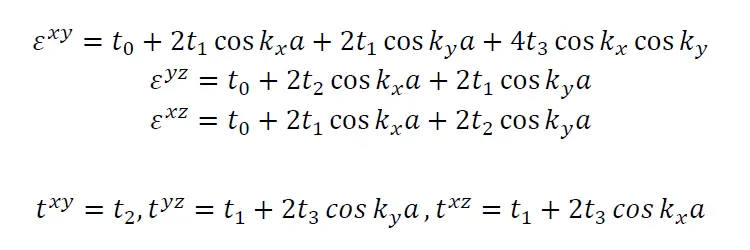
各轨道ε和t的表达式
现在我们来关注从Γ(0,0,0)点到X(0,0,pi/a)点的能带色散,yz轨道X点的能量等于tyz+εyz=(t1+2*t3)+(t0+2*t1+2*t2)=-0.416-0.114=-0.53eV。xz轨道由于对称性的原因和yz轨道的能量在这个点上简并。xy的能量等于txy+εxy=t2+t0+4t1+4t3=-0.91eV。而在Γ点,三个t2g轨道的能量是简并的,大小都等于t0+4t1+2t2+4t3。
在PHYSICAL REVIEW B 88, 125401 (2013)中,利用以上方法对SVO块体的能带进行了紧束缚近似的拟合,发现和DFT计算所得的结果非常相近。而近自由电子近似方法得到的二次型的能带(蓝线)和DFT的计算结果差异较大,特别是在X点附近的色散,能量过高。
以上,本文提供了一种基于MATLAB的wannier90hr文件的提取脚本,能高效地选取出其中重要的信息。并结合一种高对对称的体系SVO,简单解释了如何根据hr文件中的信息,运用紧束缚近似方法画出体系的能带结构。
以上内容为本人自制,谢绝转载,欢迎转发。如有错误,欢迎指正。有问题欢迎在评论栏发起讨论。
武汉格发信息技术有限公司,格发许可优化管理系统可以帮你评估贵公司软件许可的真实需求,再低成本合规性管理软件许可,帮助贵司提高软件投资回报率,为软件采购、使用提供科学决策依据。支持的软件有: CAD,CAE,PDM,PLM,Catia,Ugnx, AutoCAD, Pro/E, Solidworks 等。
 技术文档
技术文档
 推荐好文
推荐好文






 155-2731-8020
155-2731-8020




















